Researchers have shown early success creating genetically modified skin tissue to graft onto patients with a severe form of epidermolysis bullosa (EB). Stanford University Medical Center scientists conducted a phase 1 clinical trial of four adult patients who have the inherited disease, a devastating condition called recessive dystrophic EB, which causes extensive, painful blisters and skin wounds. Results of the work, which were partly supported by the NIH’s National Institute of Arthritis and Musculoskeletal and Skin Diseases, appeared recently in the Journal of the American Medical Association.
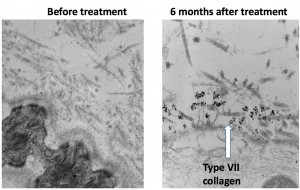
The research team took skin biopsies from patients living with recessive dystrophic EB and applied gene therapy. They used a harmless retrovirus to deliver a corrected form of the type VII collagen gene into the patients’ keratinocytes. These cells form the epidermis, or outer skin layer, and were cultured from the biopsy samples. The type VII collagen is important for binding the epidermis with the dermis, or the underlying connective tissue. Researchers coaxed the genetically modified keratinocytes to grow into sheets of skin the size of playing cards. The modified tissue was then grafted onto the patient to help speed the healing of wounds.
People who have recessive dystrophic EB cannot properly produce the type VII collagen protein due to a faulty gene. This sometimes-fatal skin disease has no cure, and supportive wound care is often the only treatment option. People with this condition are at high risk of developing aggressive skin cancer, including squamous cell carcinoma.
"Some of our patients had been dealing with wounds that had not healed for five years," said Jean Tang. M.D., Ph.D., of Stanford University, one of the study authors. "This trial shows the potential promise of gene therapy and could drastically improve the quality of life for patients living with this condition."
The researchers looked at 24 grafts applied to four patients. After three months, they took 10 biopsies from selected grafts and found the type VII collagen protein present in nine samples (90 percent). After 12 months, they found the collagen protein in five of 12 biopsy samples (42 percent). In terms of wound healing, at the three-month mark, 21 of the 24 grafts showed good progress. At the 12-month mark, 12 of 24 grafts (50 percent) were still intact. Investigators plan to continue long-term monitoring of the patients’ grafts. Hoping to build on these promising results, scientists are recruiting patients, age 13 years and older, for a phase 2 clinical trial to look at measures that include wound pain and itch.
Patients can develop a disease in which chronic wounding and scarring of the skin cause the fingers to fuse together, which results in a mitten-like appearance. "If we can stop EB in young patients, we could spare these children from experiencing chronic wounds and scarring," said Peter Marinkovich, M.D., director of the Stanford Blistering Disease Clinic and the study’s lead author.
This study was supported by the NIAMS (R01 AR055914), by funding from the Epidermolysis Bullosa Medical Research Foundation and the Epidermolysis Bullosa Research Partnership. Additional support came from the Office of Research and Development, Palo Alto VA Medical Center, and a Damon Runyon Clinical Investigator Award.
Greg Lavine
# # #
Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa.Siprashvili Z, Nguyen NT, Gorell ES, Loutit K, Khuu P, Furukawa LK, Lorenz HP, Leung TH, Keene DR, Rieger KE, Khavari P, Lane AT, Tang JY, Marinkovich MP. JAMA. 2016 Nov 1;316(17):1808-1817. doi: 10.1001/jama.2016.15588. PMID: 27802546