Two recent studies on a rare genetic disorder that causes excess bone to form have greatly contributed to our understanding of how the disease progresses, and have identified promising treatment options. The studies, funded in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), were conducted in mice and published in the Journal of Bone and Mineral Research.
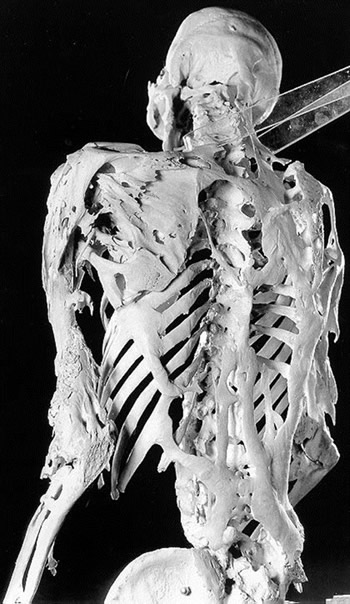
Fibrodysplasia ossificans progressiva (FOP) is a debilitating disorder that leads to heterotopic ossification (HO), an abnormal process in which extra bone develops outside the skeleton in soft tissues, such as muscles, tendons and ligaments. The disease is caused by a mutation in the ACVR1 gene, which encodes for a type I bone morphogenetic protein (BMP) receptor that promotes the formation of cartilage and bone. The mutated gene abnormally activates the BMP pathway, leading to HO episodes, also called flare-ups. The HO lesions eventually result in fused joints and loss of mobility. As the disease progresses, it can cause difficulty breathing and swallowing, and eventually death.
The excess bone can form spontaneously, but it is also spurred by an injury or trauma. An event as mild as a vaccination can cause a lesion to develop. Surgery to remove lesions is impossible because the procedure only triggers more excess bone formation and growth. Corticosteroids are sometimes used to try to quiet an HO flare, but these drugs cannot prevent it. No other treatment exists.
"FOP is a very uncommon, but devastating disease," said FOP expert Frederick Kaplan, M.D., of the Perelman School of Medicine at the University of Pennsylvania. "People with FOP essentially end up with a second skeleton that completely immobilizes them."
In one of the recent studies, Dr. Kaplan, Robert Pignolo, M.D., Ph.D., and others at the University of Pennsylvania closely observed the HO process in tissue samples from FOP patients, noting that the cells from HO lesions were profoundly lacking in oxygen, or hypoxic. This hypoxic state amplifies the ACVR1 protein’s over-activity. In lab tests, the researchers observed that by inhibiting another protein called HIF-1α—which is critical to controlling a cell’s response to hypoxia—they could dampen the wayward ACVR1 protein’s response.
The researchers then looked at several agents known to hamper HIF-1α—a compound called apigenin, which is found in parsley; the cancer drug imatinib; and an experimental agent known as PX-478. When they administered these substances to mice that were engineered to have FOP, they found that all three of the compounds deterred trauma-induced HO and protected the mice against joint immobilization.
"Knowing how critical oxygen regulation is to HO will help inform efforts to develop more effective treatments for FOP and related conditions, especially if we consider HIF-1α as a therapeutic target," said Dr. Pignolo.
The second study was conducted by a research team led by Eileen Shore, Ph.D., of the University of Pennsylvania, along with Dr. Kaplan, and Masahiro Iwamoto, D.D.S., Ph.D., and Maurizio Pacifici, Ph.D., both at the Children’s Hospital of Philadelphia. The team tested an investigational drug called palovarotene on mice engineered to carry the human FOP ACVR1 mutation. Palovarotene had been previously shown to hinder cartilage formation, a necessary step in HO formation. After feeding the mice palovarotene for 14 days, they found that the drug significantly inhibited HO formation after injury and prevented joint fusion, as well.
"If we can block injury-induced bone formation, we may be able to surgically remove existing lesions safely, which could greatly improve the quality of life for people living with this disease," said Dr. Shore.
In addition, when the investigators fed the drug to nursing female mice, offspring carrying an ACVR1 mutation were less likely to experience spontaneous HO formation. The young mice also retained near normal limb motion and normal bone growth. If the drug’s effects translate to humans, this finding suggests that human babies diagnosed with FOP could be treated before any lesions develop.
"Although we need to better understand the mechanisms through which palovarotene operates, these findings represent a promising step in establishing an effective treatment strategy for people with FOP, possibly even starting in infancy," concluded Dr. Kaplan.
Palovarotene, manufactured by Clementia Pharmaceuticals, is currently being tested in people with FOP to determine its safety and effectiveness.
This work was supported by the NIH’s NIAMS (R01-AR056837 and R01-AR041916). Additional supporters include the International Fibrodysplasia Ossificans Progressiva Association, the Center for Research in FOP and Related Disorders of the Perelman School of Medicine at the University of Pennsylvania, the Ian Cali Endowment for FOP Research, the Whitney Weldon Endowment for FOP Research, the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine, the Cali-Weldon Research Professorship for FOP Research, the Brinkman Family, the Ashley Martucci FOP Research Fund, the McGuire FOP Research Fund, the Penn Center for Musculoskeletal Disorders, and the U.S. Army Medical Research Acquisition Activity (W81XWH-07-1-0212).
Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, Mangiavini L, Groppe JC, Shore EM, Schipani E, Kaplan FS, Pignolo RJ. Bone Miner Res. 2016 Mar 30. Doi:10.1002/jbmr.2848. [Epub ahead of print.] PMID: 27027798.
Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice with the Human ACVR1R206HFibrodysplasia Ossificans Progressiva (FOP) Mutation. Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, Pacifici M, Iwamoto M, Shore EM. J Bone Miner Res. 2016 Feb 20. Doi:10.1002/jbmr.2820. [Epub ahead of print.] PMID: 26896819.
The mission of the NIAMS, a part of the U.S. Department of Health and Human Services' National Institutes of Health, is to support research into the causes, treatment and prevention of arthritis and musculoskeletal and skin diseases; the training of basic and clinical scientists to carry out this research; and the dissemination of information on research progress in these diseases. For more information about the NIAMS, call the information clearinghouse at (301) 495-4484 or (877) 22-NIAMS (free call) or visit the NIAMS website at https://www.niams.nih.gov.